TL;DR: In the US and the EU, vaccine manufacturers are required to disclose all ingredients within the vaccine on vaccine packaging inserts available online alphabetically and European public assessment reports searchable through a database, respectively. In both regions, quantities of some ingredients (active ingredients, adjuvants, and absorbents) must also be disclosed. However, the US requires disclosure of all quantities of all vaccine ingredients while the EU doesn't.
For the US, Title 21 of the Code of Federal Regulations mandates that for all biological products (which include vaccines), the packaging insert must contain information on all preservatives, antibiotics, inactive ingredients, adjuvants, and microorganisms. Packaging inserts are available directly from the FDA for all FDA approved vaccines. Thus, ingredients in vaccines cannot be kept secret. All ingredients are either of an active ingredient (microorganisms), inactive ingredient, or additive (preservatives, antibiotics, adjuvants) and must be labelled.
As an example, consider Dengvaxia, a vaccine intended to prevent dengue fever and also the most recent FDA approved vaccine1. The packaging insert is available here. All ingredients are listed under Section 11 - Description (page 13 of the linked pdf).
After reconstitution with 0.6 mL 0.4% sodium chloride, 0.5 mL of the dose contains 4.5 - 6.0 log10 CCID50 (cell culture infectious dose 50%) "of each of the chimeric yellow fever dengue (CYD) virus serotypes 1, 2, 3, and 4. Each 0.5 mL dose is formulated to contain 2 mg sodium chloride and the following ingredients as stabilizers: 0.56 mg essential amino acids (including L-phenylalanine), 0.2 mg non-essential amino acids, 2.5 mg L-arginine hydrochloride, 18.75 mg sucrose, 13.75 mg D-trehalose dihydrate, 9.38 mg D-sorbitol, 0.18 mg trometamol, and 0.63 mg urea."
In addition to just the ingredients, information on culturing of the virus is also available.
Each of the four CYD viruses ( CYD-1, CYD-2, CYD-3, and CYD-4) in DENGVAXIA was constructed using recombinant DNA technology by replacing the sequences encoding the pre-membrane (prM) and envelope (E) proteins in the yellow fever (YF) 17D204 vaccine virus genome with those encoding for the homologous sequences of dengue virus serotypes 1, 2, 3, and 4, respectively. Each CYD virus is cultured separately in Vero cells ( African Green Monkey kidney) under serum-free conditions, harvested from the supernatant of the Vero cells and purified by membrane chromatography and ultrafiltration. The purified and concentrated harvest of each CYD virus is then diluted in a stabilizer solution to produce the four monovalent drug substances. The final bulk product is a mixture of the four monovalent drug substances diluted in the stabilizer solution. The final bulk product is sterilized by filtration at 0.22 μm, filled into vials and freeze-dried.
And finally, per Title 21, we are also informed "DENGVAXIA does not contain preservative." A separate CDC regulation stipulates specification of rubber type. In this case, it is synthetic rubber.
Are there vaccine ingredients which may not be disclosed (“hidden”, “trade secret” or similar)
No, all vaccine ingredients must be disclosed in the packaging insert of the vaccine. Quantities of all ingredients must also be disclosed. This differs from the EU law (demonstrated later).
Which law defines the things that a manufacturer of a medicinal product must disclose to the authorities?
In the US, Title 21, Volume 7, Chapter I, Subchapter F, Part 610, Subpart G, Sec. 610.61 of the Code of Federal Regulations.
Was there a change in legislation or a scandal which could explain why this story emerged? Was there a gap which is maybe now closed?
The earliest version of the law I could find was in 1998. The relevant section is here on pages 23 and 24. The wording of the law appears to be extremely similar as the current version. Before 1998, the last amendment to the law was in 1990 (see the bottom of this page). Thus, the reporting requirements were in place as early as 1990 and possibly even earlier.
Title 21 of the Code of Federal Regulations
The following items shall appear on the label affixed to each package containing a product:
The preservative used and its concentration, or if no preservative is used and the absence of a preservative is a safety factor, the words "no preservative";
The type and calculated amount of antibiotics added during manufacture;
The inactive ingredients when a safety factor, or reference to an enclosed circular containing appropriate information;
The adjuvant, if present;
The source of the product when a factor in safe administration;
The identity of each microorganism used in manufacture, and, where applicable, the production medium and the method of inactivation, or reference to an enclosed circular containing appropriate information;
According to Law.SE, all inactive ingredients are listed. Additionally, I have contacted the FDA who provided similar information as I have already outlined in the answer.
For the EU, Article 59 of Title V of Directive 2001/83/EC states (page 42 of the linked pdf):
The package leaflet shall be drawn up in accordance with the summary of the product characteristics; it shall include, in the following order:
Thus, all the name and quantity of all components of the vaccine must be published in the EU as well.
The European Medicines Agency has a separate document titled "Guideline on quality aspects included in the product information for vaccines for human use". Page 4 reads
The principal entries under Section 2 in the SmPC [summary of product characteristics] should appear in the following order:
- Qualitative and quantitative declaration of each active substance,
- Qualitative and quantitative declaration of any adjuvant or adsorbant present,
- Origin of the active substance, if applicable,
- Residues of clinical relevance, if applicable,
- Excipients with known effects, if applicable,
- a reference to the full list of excipients in 6.1,
Excipients are all components of a vaccine that are not active ingredients (e.g. preservatives, adjuvants, and stabilizers). As the quantity, type, and origin of active ingredients must be disclosed, we can conclude that the EU requires labelling of all ingredients within the vaccine. The linked pdfs also contain other more specific labeling guidelines.
The earliest version of the law that I could find was in 2001. The requirement to disclose all ingredients was still there (see page 36 and 37 of the linked pdf). However, I noticed fewer guidelines on reporting the risks of excipients (e.g. In 2001, manufacturers had to report all excipients, but manufacturers didn't have to report the known risks; now manufacturers have to do both).
In the EU, the European Medicines Agency is responsible for approving vaccines. A database of all approved medicines is here. A search specifically for human vaccines is here. As an example, consider the most recently approved non-flu vaccine in the EU, which is also Dengvaxia (authorized 12/12/2018). The authorization website contains various documents, including a 64 page European public assessment report. The report contains a list of excipients in Section 6.1 (page 15).
Powder:
Solvent:
Sodium chloride
Water for injections
At first glance the sections of the report appears to be largely duplicated (e.g. pages 2 through 16 are largely duplicated again in pages 17 through 32). Confusingly, this is because half of it apply to Dengvaxia in pre-filled syringe while the other half apply to Dengvaxia in multidose containers.The packaging leaflet for the user (pages 49 through 56) contains the vaccine ingredients as well (page 53).
After reconstitution, one dose (0.5 mL) contains 4.5 - 6.0 log10 CCID50* of each serotype of the chimeric yellow fever dengue virus** (1, 2, 3 and 4) (live, attenuated).
- CCID50: 50% Cell Culture Infectious Dose.
- Produced in Vero cells by recombinant DNA technology. This product contains genetically modified organisms (GMOs).
The other ingredients are: essential amino acids including Phenylalanine, non-essential amino acids, Arginine hydrochloride, Sucrose, Trehalose dihydrate, Sorbitol (E420), trometamol, urea, sodium chloride, water for injections.
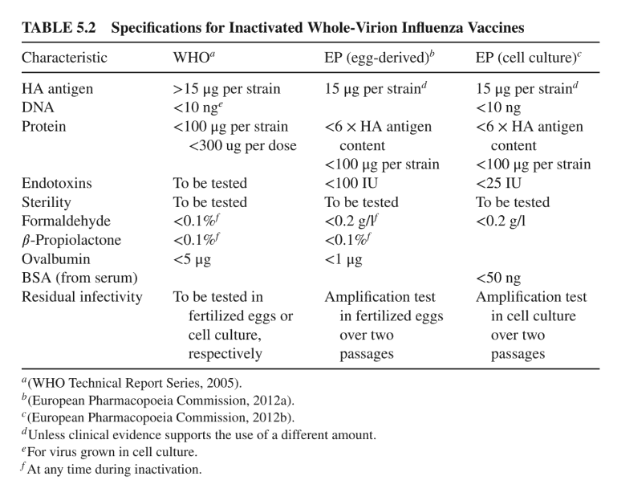
The reason I am listing this out is to show a major difference between EU and US laws. This is reflected in the European public assessment report (from the EU) and the packaging insert (from the US FDA). Excipients are listed for both situations but the concentration and volume (e.g. quantitative descriptions) are only listed for the packaging insert. The EU law requires disclosure of quantities of active ingredients, adjuvants, and absorbents. If an excipient is neither an adjuvant or absorbent (such as sodium chloride) the concentration and volume are not disclosed. This is reflected in the European public assessment report.
Are there vaccine ingredients which may not be disclosed (“hidden”, “trade secret” or similar)
No, all vaccine ingredients must be disclosed in the summary of product characteristics of the vaccine. Quantities of active ingredients, adjuvants, and absorbents must also be disclosed. Quantities of other ingredients need not be disclosed (and usually aren't). This is the biggest difference between the EU and the US laws.
Which law defines the things that a manufacturer of a medicinal product must disclose to the authorities?
In the EU, Article 59 of Title V of Directive 2001/83/EC.
Was there a change in legislation or a scandal which could explain why this story emerged? Was there a gap which is maybe now closed?
The earliest version of the law I could find was in 6 November 2001. I suspect this was the very first version of the law as the directive is "2001/83." The reporting requirements were in place as early as 2001.
Note regarding the textbook image: The textbook mentioned in the claim in the question does indeed exist. Additionally, the underlined sentence is real. We can check this on Google Books. Page 77 of the book clarifies this.
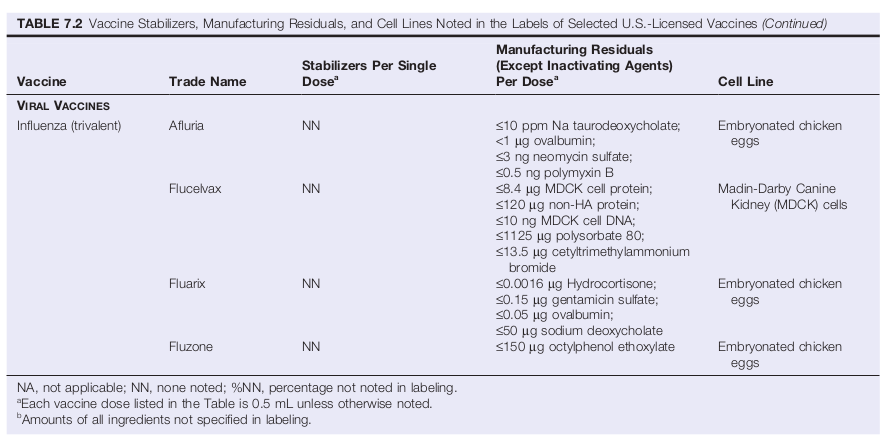
The Food, Drug and Cosmetic Act (Section 502[e][1][A][iii]) states that all inactive ingredients should be noted in labeling; it also states that this requirement is not necessary if trade secret information would be disclosed. The CFR additionally notes that an inactive ingredient should be listed in the labeling if the ingredient's presence is considered a safety factor (21 CFR 610.61[n]). In some cases, even in the absence of any evidence that a particular material might pose a safety factor, manufacturers have elected to disclose the presence of residual materials such as detergents, solvents, and chelating agents (see Table 6-2 for examples of manufacturing residuals).
The CFR law has been quoted earlier. Following is the relevant excerpt from the Food, Drug, and Cosmetic Act (FD&C Act).
(iii) the established name of each inactive ingredient listed in alphabetical order on the outside container of the retail package and, if determined to be appropriate by the Secretary, on the immediate container, as prescribed in regulation promulgated by the Secretary, except that nothing in this subclause shall be deemed to require that any trade secret be divulged, and except that the requirements of this subclause with respect to alphabetical order shall apply only to nonprescription drugs that are not also cosmetics and that this subclause shall not apply to nonprescription drugs not intended for human use.
As I just dug up the textbook ~20 minutes ago, I do not have enough time to resolve this apparent contradiction in law. Potentially, the CFR and the FD&C Act can conflict when a trade secret prevents disclosure of an ingredient. I have asked this on Law.SE and will update this answer later.
My interpretation is: If the trade secret impacts safety, it must be disclosed. If the trade secret does not impact safety, it does not have to be disclosed. As such, the quote from the textbook "Some information regarding additives and residuals is considered to be a trade secret and thus confidential, and cannot be discussed in this chapter." is taken out of context. Such trade secrets can only be kept when there is no impact on safety. Again, I will update this answer later.
1Headline under FDA Official Releases: "FDA approves Dengvaxia, the first vaccine approved for the prevention of dengue disease in people ages 9 through 16 who have laboratory-confirmed previous dengue infection and who live in endemic areas." For those curious and interested, there is another news release here titled "First FDA-approved vaccine for the prevention of dengue disease in endemic regions."